

0 引言
【研究意义】薰衣草(Lavandula angustifolia Mill)是唇形科薰衣草属多年生小灌木植物。我国薰衣草主要分布于新疆伊犁河谷,该河谷薰衣草种植面积占全国的98%。薰衣草精油具有较广泛的商业价值[1-2]。而细菌作为土壤微生物的主要成分,具有分解、矿化和促进土壤物质循环和能量流动等多种生态功能,在调节土壤肥力、植物健康等方面发挥着关键作用。由于其对环境变化十分敏感,通常作为评估土壤养分循环和维持生态系统平衡的重要指标[3-4]。探究不同连作年限薰衣草根际土壤细菌多样性及其群落组成对了解其生长环境的变化及科学管理具有重要意义。【前人研究进展】目前,对薰衣草的相关研究诸多,例如其产物精油如对抑菌作用的探究等[5
1 材料与方法
1.1 材料
研究区域选在新疆伊犁哈萨克自治州霍城县 (43°39'~ 44°50'N,80°11'~24'E) 新疆生产建设兵团六十六团薰衣草种地。霍城县是我国最大薰衣草种植基地,年均气温为8.2~ 9.4℃,年日照时平均数为2 826 h,全年降水量为140~ 460 mm,海拔在 760 m左右,土质松软,含砂质,略偏碱性,适宜薰衣草生长[17]。土样采自同一生态区域,于2019年7月初薰衣草旺盛期进行采样。
1.2 方法
1.2.1 试验设计
样本共有4组,即未种植薰衣草土样 (L0作空白对照组)、种植薰衣草1年、3年及5年土样(分别为L1、L3和L5作试验组)。空白对照组土样采自离薰衣草田地50~ 150 m处空地土壤。每组样品均设3个重复。每个样地采用多点采样法,采集样地5~ 10 cm深处的土壤。各样品分别混匀后放入冰盒内带回实验室。经2 mm钢筛去杂,充分混匀装袋存储于-80℃冰箱备用。
1.2.2 土壤细菌16S rDNA序列的 PCR 扩增
采用土壤 DNA 提取试剂盒提取12个混合样本土壤微生物基因组 DNA,以稀释后的微生物基因组 DNA 为模板,利用细菌核糖体16S rDNA 区引物515F(5'-GTGCCAGCMGCCGCGGTAA-3')和806R(5'-GGACTACHVGGGTWTCTAAT-3')进行 PCR 扩增。PCR反应体系:基因组 DNA (0.1 μL) 10~ 100 ng,正、反向引物(10~ 20 pmol)各 2 μL,10×PCR Buffer 5 μL,Mg 2+(25 mmol/L)3 μL,dNTP(each 10 mmol/L)1 μL,ddH2O 补齐到 50 μL。PCR 反应条件:95℃ 3min;95℃ 30 s,55℃ 30 s,72℃ 1 min,30个循环;72℃ 5 min。扩增后的PCR 产物进行2% 琼脂糖凝胶电泳检测,并对扩增后的土壤基因组 DNA 进行高通量测序。测序工作由北京诺禾致源生物信息科技有限公司完成。
1.3 数据处理
2 结果与分析
2.1 测序结果质量比较
研究表明,通过对薰衣草根际土壤样品测序得到原始序列条数为1 075 923,过滤掉低质量的序列后,共获得有效序列总数为793 563,平均长度为253 bp。根据 barcode 标签进行样品序列拆分,对初始序列进行去冗余处理后,获得16S rRNA Unique Reads,并将其聚类为用于物种分类的OTU,统计得到各个样品在不同 OTU 中的绝对丰度信息,OTU 个数越多该样品丰富度越高。所有样本Q30 值均高于96% 。同时每个样本Q30碱基百分比均在96%~98%,过滤后样本均一性较好。L0、L1、L3 和 L5 样本中有效序列分别为66 833、68 385、63 631和64 800个,且除 L3样本 OTU 数量略低,而覆盖度略高外,各样本组间不存在其他显著性差异(P<0.05)。表1
表1 样品测序数据统计
Tab.1
| 样本 Sample | 原始序列数 Raw tags | 有效序列数 Effective tags | 有效序列比例 Effective(%) | OTU数量 Number of OTU | 覆盖度 Goods-coverage |
|---|---|---|---|---|---|
| L0 | 95 166±11 883a | 66 833±2 505a | 71.12±7.26a | 3 701±42a | 0.990ab |
| L1 | 90 524±9 124a | 68 385±2 469a | 77.07±9.45a | 3 869±218a | 0.990a |
| L3 | 91 256±225a | 63 631±2 447a | 69.56±2.85a | 3 345±140b | 0.991±0.001b |
| L5 | 84 562±6 163a | 64 800±2 678a | 77.4±5.98a | 3 810±149a | 0.989a |
注:不同字母表示不同连作年限之间的差异显著(P<0.05),下同
Notes:Different letters indicate significant difference between different years of continuous cropping(P<0.05),the same as below
2.2 细菌OTUs分析
研究表明,不同连作年限样品之间存在共OTUs数量为2 859个,约占总数的44.75%,近50% OTUs 所对应的细菌物种不易受到连作年限的影响,对环境的适应性较强。L0、L1、L3和L5四组样品特有 OTU 数量分别为 257、275、291 和 228,而各组样品总 OTU 数量分别为 4 588、4 897、4 269 和 4 804,各组特有 OTU 数占其总 OTU 数量比例分别为 5.60 %、5.62 %、6.82 %和 4.75 %,不同种植年限薰衣草根际细菌多样性存在差异,且种植薰衣草第3年土壤中特有物种激增。在不计对照组L0 情况下,L1、L3 和 L5 三组样品特有 OTU 数量分别为 532、410 和 447,占各组总OTU 数量的比例分别为 10.86%、9.60%和 9.30%,随着连作年限的增加,土壤特有物种在逐渐减少。图1
图1
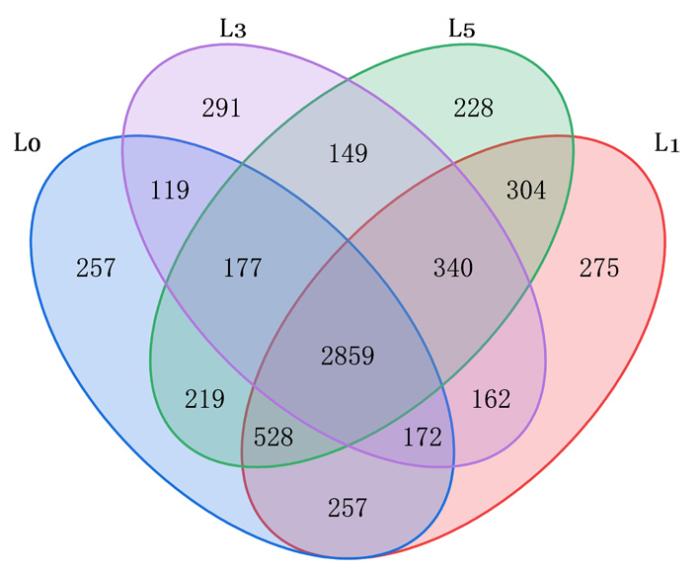
图1
基于OTU水平的土壤样本中细菌群落的Venn图
Fig.1
Venn plot of bacterial communities in soil samples based on OTU level
2.3 细菌群落Alpha多样性
研究表明,细菌群落多样性变化规律从大到小依次为L0> L1> L5> L3,细菌群落多样性随着种植年限的增加先减少后增加,且种植薰衣草土壤细菌群落多样性均低未种植土壤,薰衣草的种植降低了土壤细菌多样性,第3年尤其明显,后虽有所回升却仍有差距。细菌群落丰富度随着种植年限的增加仍呈先减后增的变化趋势,其变化规律从大到小依次为 L1> L5> L0> L3,Shannon指数在L3与L0、L1两组间具有明显的差异(P值分别为0.009 4、0.049 3),与L5无明显差异(P>0.05);与此同时Chao1指数中L3与L1、L5两组也存在显著差异,而与L0不显著(P值依次为0.010 5、0.017、0.057 8)。种植年限为3年时,土壤微生物群落的丰富度和多样性均和其他年限有一定区别。表2
表2 样本细菌的Alpha多样性指数
Tab.2
| 样本 Sample | Shannon指数 Shannon index | Simpson指数 Simpson index | Chao1指数 Chao1 index | Ace指数 Ace index |
|---|---|---|---|---|
| L0 | 9.801±0.113ac | 0.997±0.001a | 3 635.526±43.744ab | 3 726.847±67.421a |
| L1 | 9.680±0.267ac | 0.997±0.002ab | 3 854.589±342.052b | 3 907.551±395.014a |
| L3 | 9.282±0.357b | 0.995±0.002b | 3 359.662±184.162a | 3 387.528±146.537b |
| L5 | 9.639±0.025bc | 0.996±0.001ab | 3 757.002±138.369b | 3 815.712±163.43a |
2.4 细菌群落组成
研究表明,归类共检测到 48 个门,53 个纲,127 个目,253 个科,562 个属等分类类群。
2.4.1 基于门水平的细菌群落组成
研究表明,有10个细菌门的平均相对丰度超过1%。根际土壤样本的中心优势细菌群落主要由酸杆菌门(Acidobacteria) 、变形菌门(Proteobacteria) 、放线菌门(Actinobacteria) 、拟杆菌门(Bacteroidetes) 4个门类组成。酸杆菌门、放线菌门、芽单胞菌门、浮霉菌门和疣微菌门相对丰度随着连作年限的增加先增加后减少,酸杆菌门相对丰度依次为23.58%、31.31%和23.41%,放线菌门相对丰度依次为6%、7.40%和7.20%,芽单胞菌门相对丰度依次为5.82%、6.35%和5.11%,浮霉菌门相对丰度依次为2.97%、5.80%、和3.86%,疣微菌门相对丰度依次为4.46%、5.96%和5.55%。而变形菌门、拟杆菌门、厚壁菌门和绿弯菌门的相对丰度则是先减少后增加,依次分别为32.03%、25.83%、31.36%;9.16%、6.57%、9.15%;5.90%、0.72%和4.65%。奇古菌门的相对丰度则逐年下降但变化不大,分别为1.92%、1.91%和1.86%。与未种植薰衣草的土壤相比,放线菌门和厚壁菌门的相对丰度明显降低,而酸杆菌门、变形菌门和奇古菌门相对丰度明显增高,薰衣草的种植在一定程度上直接或间接对放线菌门和厚壁菌门生长具有抑制作用,但有利于后三者的生长繁殖,基于门水平的细菌群落成员受到调控。未被分类的细菌比例较低,均低于6.1%。图2
图2
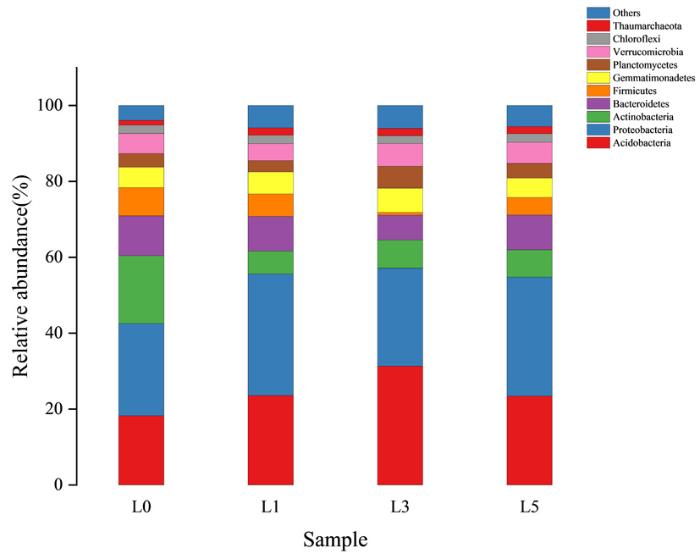
2.4.2 基于科水平的细菌群落组成
研究表明,在科水平上种植薰衣草样本组中共分类到146个类群,未被分类细菌占60.82%~70.40%。其中酸杆菌门中未被分类的一个科以及芽单胞菌科(Gemmatimonadaceae)以及未知中文名称(Pyrinomonadaceae)为优势科,且酸杆菌门中未被分类的一个科在种植薰衣草后相对丰度显著下降,在第5年又恢复甚至高于原本水平,该菌种的生长在种植初期受到严重抑制,随着时间的推移逐渐适应了环境的变化回到正常水平。此外,鞘脂单胞菌科(Sphingomonadaceae)在第5年基本恢复未种植水平,亚硝化单胞菌科(Nitrosomonadaceae)在种植薰衣草后明显增多。综上,从种植薰衣草的第1年起未种植薰衣草土壤原有微生物群落发生一定变化,其优势菌相对丰度或下降或上升,原本核单胞菌科等相对丰度不占优势的科转变为优势菌科。在种植薰衣草3组土样中肠杆菌科(Enterobacteriaceae)、微颤蓝细菌科(Microscillaceae)、伯克氏菌科等类群表现为先下降后上升的变化趋势,种植年限3年的土样中的相对丰度最低;而芽单胞菌科(Gemmatimonadaceae)、噬几丁质菌科等类群与此相反,相对丰度表现为先上升后下降的趋势,种植年限3年的土样中相对丰度最高。图3
图3
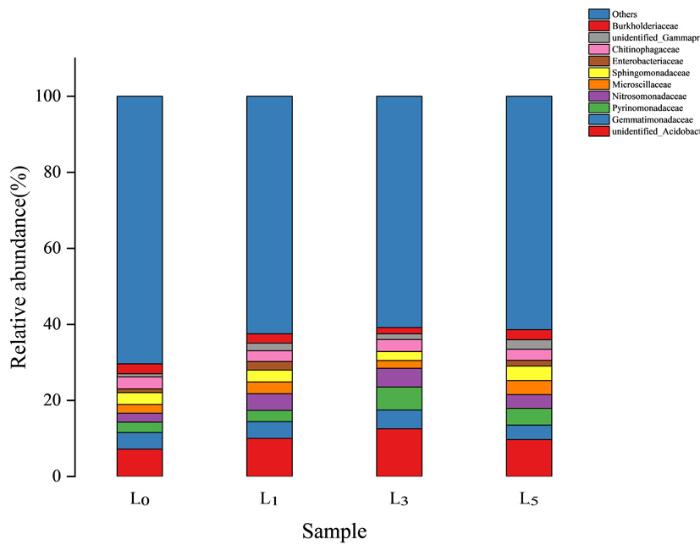
2.4.3 基于属水平的细菌群落组成
研究表明,在属水平上3组种植薰衣草样本组中共划分为562个类群,其中平均相对丰度均大于1% 的有酸杆菌门中未被分类的一个属、鞘氨醇单胞菌属(Sphingomonas)和嗜酸性杆菌属(Stenotrophobacter)等3个属,而未知菌类占86.26%~89.65%,土壤生态系统仍是值得我们继续探索的微生物宝库。在种植薰衣草的土样中酸杆菌门中未被分类的一个属仍为最优势细菌,所占比例分别为5.73%、5.96%、5.42%,均高于未种植土壤(3.18%)。而其他优势菌则略有不同,除上述菌种外在种植年限为1年土样中依次为鞘氨醇单胞菌属(1.86%)>不动杆菌属(Acinetobacter,1.11%)>嗜酸性杆菌(1.10%),在种植年限为3年土样中依次为嗜酸性杆菌(1.41%)>鞘氨醇单胞菌属(1.14%),在种植年限为5年土样中依次为鞘氨醇单胞菌属(1.71%)>类固醇杆菌属(1.44%)>嗜酸性杆菌(1.29%)>肠杆菌科未被分类的一个属(1.09%)>新鞘脂菌属(Novosphingobium)(1.01%),其中不动杆菌属、类固醇杆菌属、肠杆菌科未被分类的一个属和新鞘脂菌属占比均呈升高、降低再升高的趋势。对照组中西索恩氏菌属、黄杆菌属(Flavobacterium)的相对丰度显著高于各种植组(>1%),而金线藻属(Chryseolinea)与之相反,相对丰度均不足1%。图4
图4

2.5 细菌群落Beta多样性
研究表明,土壤样品可被分为三个类组,其中L0样本为一组,L1和L5样本为第二组,L3样本为第三组。3组在MDS1和MDS2轴上明显相互分离,未种植与种植薰衣草土壤中细菌群落存在较大差异,且种植年限为1年和5年的土壤细菌组成具有相当程度的相似性。而种植年限为3年与上述二者距离较远,反映其土壤细菌群落与种植了1年和5年的土壤有较大的差异。共分为两个大支,即种植薰衣草和未种植薰衣草两部分,前者又分为两个支,L1、L5为一支,L3为另一支。薰衣草的种植明显改变了土壤细菌群落的Beta多样性。图5
图5

图5
细菌非度量多维尺度图(a)和基于Beta多样性距离的样品层级聚类树(b)
Fig.5
Bacterial NMDS diagram(a)and sample hierarchical clustering tree based on Beta diversity distance
3 讨论
3.1 物种积累箱线图和和物种稀疏曲线随着样本数的增加趋于达到饱和平台,表明采样强度和测序深度足够[20]。以97%的一致性将序列聚类成为OTUs,共得到6 389个OTUs,再利用 Origin 软件绘出韦恩图,分析不同样本组之间共有、特有OTU数目,直观展示样品间OTU的重叠情况[21]。Shannon 指数越大细菌群落多样性越高,Simpson 指数值越大,说明群落多样性越低[22],种植3年的薰衣草土壤细菌群落丰富度低于未种植土壤,此时的土壤微生物生态发生紊乱[23]。土壤微生物作为土壤的重要组成部分和土壤养分循环的主要推动者,其群落结构的变化在一定程度上可以反映土壤质量的变化情况[24]。土壤细菌群体量大、种类多,是检测土壤质量变化的重要指标,在评价土壤生态系统等功能方面具有重要作用[25]。土壤-微生物-植物间的互作一直是土壤微生物研究的热点方向之一,其互作关系的复杂性影响整个根际系统,连作障碍就是由这种复杂互作关系引起的,单一作物连续多年种植导致根际分泌物及残体的逐年积累,使土壤特性尤其是土壤微生物组发生变化导致产量下降,造成土壤微生物区系紊乱[26,27]。Tan等[28]发现根际微生态系统的失衡导致病原菌大量繁殖的同时有益菌的生长也受到抑制,造成植物生长发育不良,影响产量及品质形成。
3.2 高通量测序技术可用于对土壤微生物群落的DNA特定片段进行高质量的测序[29]。研究采用16S rRNA Illumina 高通量测序技术分析了连作种植对薰衣草根际土壤细菌群落结构和多样性的影响。4个样本组(共12个样本)均采自同一种植地,且相互距离较近,所对应的环境因素基本相同,如气候、温度、降雨量等,能比较客观地反映出连作年限对根际土壤细菌多样性的影响。Chao1 指数与Shannon 指数的变化规律说明薰衣草根际土壤细菌群落相对丰度与多样性均呈先减后增的变化趋势,种植年限1年的土壤中最高,这可能与种植初期土壤微生态系统尚不稳定,仍处于恢复状态有关,与Xue等[30]采用变性梯度凝胶电泳技术鉴定的5、12和25 年生观赏牡丹根际土壤细菌群落结构变化结果并不相同,可能与植被类型、生境的土壤特性以及连作年限不同有关;在种植3年的土壤中最低,且OTU数量也显著减少,微生态系统此时再次发生较大变化。每组薰衣草土壤样品特有OTU 数量占该组总OTU 数量的比例较高,说明种植年限对根际土壤细菌群落演替影响较大,并随着种植年限的增加,根际土壤特有物种逐渐减少,可能是因为部分细菌不适宜土壤环境的变化逐渐减少甚至消失。
3.3 研究亦表明薰衣草土壤细菌群落结构变化较小,但也因种植年限不同而存在一定程度的差异。在门水平上,薰衣草土壤中优势菌门为酸杆菌门、变形菌门、放线菌门和拟杆菌门4个类群。与以往对土壤中细菌群落的研究结果基本一致[5,31,32],但也存在不同情况。如杨宝钰[33]研究甜高粱秸秆发现门水平相对丰度最高为厚壁菌门。研究发现酸杆菌门在酸性土壤中含量更高,其丰度与pH水平呈负相关[34-
3.4 在测序样本相同的基础上,陈雪静等[45]研究发现真菌群落多样性和群落结构的变化程度相对较大,属水平上的优势菌株均为致病菌,球囊霉属的OUT数量随种植年限的延迟逐渐减少。微生物群落对环境变化的响应机制依然不明确,仍需要我们继续探索。土壤微生物群落功能指标与土壤理化性质关系密切,相互影响[16],土壤有机碳和有效磷含量是影响细菌群落结构变化的重要因素。烟草连作种植实验采样测序结果显示没有连作障碍样本的土壤有机质(SOM)、有效磷(AP)、总碳(TC)、硝酸根含量均显著高于存在连作障碍的样本[46]。与单一连作同种植物相比,轮作和间作更有利于植物的生长[47-
4 结论
随着连作年限的增加,Shannon指数和Chao1指数均先下降后上升。在种植薰衣草的土壤细菌群落中,在门水平上,酸杆菌门(Acidobacteria) 、变形菌门(Proteobacteria) 、放线菌门(Actinobacteria)是优势门,且酸杆菌门和放线菌门的相对丰度随年限先减少后增加,而变形菌门先增加后减少。在属水平上优势菌属为酸杆菌门中未被分类的一个属、鞘氨醇单胞菌属(Sphingomonas)和嗜酸性杆菌属(Stenotrophobacter),鞘氨醇单胞菌属的相对丰度先减少后增加,而酸杆菌门中未被分类的一个属以及嗜酸性杆菌属先增加后减少,优势微生物群落随着时间的推移发生了一定的变化。与未种植的土壤相比,种植后的细菌群落也表现出显著变化,第1年与第5年相似,与第3年相比有显著差异。持续种植薰衣草会引起土壤微生物群落结构和多样性的变化。
参考文献
Current trends for lavender (Lavandula angustifolia mill.) crops and products with emphasis on essential oil quality
[J].
Effects of nanoparticles treatments and salinity stress on the genetic structure and physiological characteristics of Lavandula angustifolia Mill
[J].
Comparative analysis of bacterial community structure in the rhizosphere of maize by high-throughput pyrosequencing
[J].
Soil microbial community and its interaction with soil carbon and nitrogen dynamics following afforestation in Central China
[J].
薰衣草精油通过抑制凋亡、上调海马BDNF/proBDNF表达改善大鼠卒中后抑郁
[J].
Lavender essential oil improves post-stroke depression in rats by inhibiting apoptosis and up-regulating BDNF/proBDNF expression in hippocampus
[J].
Objective To explore the effect of lavender essential oil on the expression of brain-derived neurotrophic factor (BDNF)/pro-brain-derived neurotrophic factor (proBDNF) and the recovery of depression in the ischemic hippocampus of rats after stroke. <br>Methods A total of 45 male SD rats were randomly divided into a control group, a model group, and an intervention group, with 15 rats in each group. Rats in the control group raised under conventional conditions were subjected to intraperitoneal injection of physiological saline as a control experiment. In the model group, middle cerebral artery occlusion/reperfusion (MCAO/r) model was established and rats were given intraperitoneal injection of physiological saline. In the intervention group, rats were given lavender essential oil and exercise intervention after MCAO/r. The anxiety and depression behaviors of rats were tested through outdoor venues, forced swimming, and sucrose preference. The levels of oxidative stress biomarkers in rats were detected using commercial kits, and the expression of BDNF/proBDNF signaling pathway was analyzed by reverse transcription-polymerase chain reaction (RT-PCR). The ratio of BDNF/proBDNF in CA1 region, CA3 region and dentategyrus (DG) regions of ischemic hippocampal tissue was analyzed by Western blotting (WB). The correlation analysis was performed between the BDNF/proBDNF ratio and behavioral tests for depression. Neurobehavioral (sensory and motor) testing was used to detect neural defects in rats. <br>〖MM(〗〖HT6SS〗〖CM10-8〗河北医科大学学报第44卷第11期〖MM)〗<br>Results The stationary time of the model group was longer than that of the control group, and the sucrose preference, climbing frequency, and exercise distance were lower than those of the control group. The static time of the intervention group was longer than that of the control group and shorter than that of the model group, while the sucrose preference, climbing frequency, and exercise distance were lower than those of the control group and higher than those of the model group (P<0.05). The levels of superoxide dismutase (SOD), glutathione,r-glutamyl cysteingl + glycine (GSH) and fluoride resistant acid phosphatase (FRAP) in the model group were lower than those in the control group, and the levels of malondialdehyde (MDA) were higher than those in the control group. The levels of SOD, GSH and FRAP in the intervention group were higher than those in the model group, while the level of MDA was lower than that in the control group and the model group (P<0.05). The mRNA expressions of BDNF, neurotrophic receptor (TrkB) and microtubule-associated protein (DCX) in the model group were lower than those in the control group, while the mRNA expressions of proBDNF and neurotrophic receptor P75 (p75NTR) were higher than those in the control group. The mRNA expressions of BDNF, TrkB, and DCX in the intervention group were lower than those in the control group and higher than those in the model group, while the mRNA expressions of proBDNF and p75NTR were higher than those in the control group but lower than those in the model group (P<0.05). The BDNF/proBDNF ratio in the CA1, CA3, and DG regions of the model group was lower than that of the control group, which was higher in the intervention group than in the model group (P<0.05). The BDNF/proBDNF ratio was negatively correlated with resting time during forced swimming (r=-0.74, P<0.01), while the BDNF/proBDNF ratio was positively correlated with sucrose preference (r=0.63, P<0.01). The neurological deficit score in the model group was higher than that in the control group, which was higher in the intervention group than in the control group and lower than that in the model group (P<0.05). The expression of Bcl-2 related X protein (Bax)/B-cell lymphoma-2 gene (Bcl-2) in the model group was higher than that in the control group, while the expression of Bcl-2 was lower than that in the control group. The expression of Bax and Bax/Bcl-2 in the intervention group was lower than that in the model group, while the expression of Bcl-2 was higher than that in the model group (P<0.05). <br>Conclusion Lavender essential oil may improve neurological function, enhance endogenous antioxidant defense, inhibit oxidative stress pathway and nerve apoptosis by regulating the relative level of BDNF and proBDNF, and then improve the depressive behavior of PSD rats.<br><div> <br></div>
Comparison of Lavandula angustifolia mill. tincture and imipramine in the treatment of mild to moderate depression: a double-blind, randomized trial
[J].
5种植物精油抗菌性能研究及特征成分分析
[J].
Investigation on antibacterial properties and characteristic components of five kinds of plant essential oils
[J].
伊犁州薰衣草根际促生真菌的分离鉴定及活性探究
[J].
Isolation,Identification and Activity Research of lavender rhizosphere growth-promoting fungi in Yili State
[J].
Exploring the chemical composition of Bulgarian lavender absolute (Lavandula angustifolia mill.) by GC/MS and GC-FID
[J].
Biotechnologically produced Lavandula angustifolia mill. extract rich in rosmarinic acid resolves psoriasis-related inflammation through Janus kinase/signal transducer and activator of transcription signaling
[J].
Intercropping with garlic alleviated continuous cropping obstacle of cucumber in plastic tunnel
[J].
Rhizosphere microorganisms and soil physicochemical properties of restored wetland plant communities at cutting slash of Populus deltoides in Dongting Lake
[J].
UPARSE: highly accurate OTU sequences from microbial amplicon reads
[J].Amplified marker-gene sequences can be used to understand microbial community structure, but they suffer from a high level of sequencing and amplification artifacts. The UPARSE pipeline reports operational taxonomic unit (OTU) sequences with ≤1% incorrect bases in artificial microbial community tests, compared with >3% incorrect bases commonly reported by other methods. The improved accuracy results in far fewer OTUs, consistently closer to the expected number of species in a community.
Bacterial diversity in aquatic and other environments: what 16S rDNA libraries can tell us
[J].We evaluate the substantial amount of information accumulated on bacterial diversity in a variety of environments and address several fundamental questions, focusing on aquatic systems but including other environments to provide a broader context. Bacterial diversity data were extracted from 225 16S rDNA libraries described in published reports, representing a variety of aquatic and non-aquatic environments. Libraries were predominantly composed of rare phylotypes that appeared only once or twice in the library, and the number of phylotypes observed was correlated with library size (implying that few libraries are exhaustive samples of diversity in the source community). Coverage, the estimated proportion of phylotypes in the environment represented in the library, ranged widely but on average was remarkably high and not correlated with library size. Phylotype richness was calculated by methods based on the frequency of occurrence of different phylotypes in 194 libraries that provided appropriate data. For 90% of aquatic-system libraries, and for 79% of non-aquatic libraries, the estimated phylotype richness was <200 phylotypes. Nearly all of the larger estimates were in aquatic sediments, digestive systems and soils. However, the approaches used to estimate phylotype richness may yield underestimates when libraries are too small. A procedure is described to provide an objective means of determining when a library is large enough to provide a stable and unbiased estimate of phylotype richness. A total of 56 libraries, including 44 from aquatic systems, were considered 'large enough' to yield stable estimates suitable for comparing richness among environments. Few significant differences in phylotype richness were observed among aquatic environments. For one of two richness estimators, the average phylotype richness was significantly lower in hyperthermal environments than in sediment and bacterioplankton, but no other significant differences among aquatic environments were observed. In general, and with demonstrated exceptions, published studies have captured a large fraction of bacterial diversity in aquatic systems. In most cases, the estimated bacterial diversity is lower than we would have expected, although many estimates should be considered minimum values. We suggest that on local scales, aquatic bacterial diversity is much less than any predictions of their global diversity, and remains a tractable subject for study. The global-scale diversity of aquatic Bacteria, on the other hand, may be beyond present capabilities for effective study.
Precipitation exerts a strong influence on arbuscular mycorrhizal fungi community and network complexity in a semiarid steppe ecosystem
[J].
Venn diagrams in bioinformatics
[J].
Effect of organic substitution rates on soil quality and fungal community composition in a tea plantation with long-term fertilization
[J].
A green garlic (Allium sativum L.) based intercropping system reduces the strain of continuous monocropping in cucumber (Cucumis sativus L.) by adjusting the micro-ecological environment of soil
[J].
高通量测序技术分析新疆新源县过度放牧土壤细菌多样性
[J].
Determination of bacteria diversity of degraded grassland in Xinyuan county by high-throughput sequencing technology
[J].
Soil bacterial and fungal community dynamics in relation to Panax notoginseng death rate in a continuous cropping system
[J].
Growth and physiological changes in continuously cropped eggplant (Solanum melongena L.) upon relay intercropping with garlic (Allium sativum L.)
[J].
Root exudates: from plant to rhizosphere and beyond
[J].This article describes the composition of root exudates, how these metabolites are released to the rhizosphere and their importance in the recruitment of beneficial microbiota that alleviate plant stress. Metabolites secreted to the rhizosphere by roots are involved in several processes. By modulating the composition of the root exudates, plants can modify soil properties to adapt and ensure their survival under adverse conditions. They use several strategies such as (1) changing soil pH to solubilize nutrients into assimilable forms, (2) chelating toxic compounds, (3) attracting beneficial microbiota, or (4) releasing toxic substances for pathogens, etc. In this work, the composition of root exudates as well as the different mechanisms of root exudation have been reviewed. Existing methodologies to collect root exudates, indicating their advantages and disadvantages, are also described. Factors affecting root exudation have been exposed, including physical, chemical, and biological agents which can produce qualitative and quantitative changes in exudate composition. Finally, since root exudates play an important role in the recruitment of mycorrhizal fungi and plant growth-promoting rhizobacteria (PGPR), the mechanisms of interaction between plants and the beneficial microbiota have been highlighted.
Rhizospheric soil and root endogenous fungal diversity and composition in response to continuous Panax notoginseng cropping practices
[J].
Overview of next-generation sequencing technologies
[J].
Changes in soil microbial community structure with planting years and cultivars of tree peony (Paeonia suffruticosa)
[J].
Prokaryotes: the unseen majority
[J].The number of prokaryotes and the total amount of their cellular carbon on earth are estimated to be 4-6 x 10(30) cells and 350-550 Pg of C (1 Pg = 10(15) g), respectively. Thus, the total amount of prokaryotic carbon is 60-100% of the estimated total carbon in plants, and inclusion of prokaryotic carbon in global models will almost double estimates of the amount of carbon stored in living organisms. In addition, the earth's prokaryotes contain 85-130 Pg of N and 9-14 Pg of P, or about 10-fold more of these nutrients than do plants, and represent the largest pool of these nutrients in living organisms. Most of the earth's prokaryotes occur in the open ocean, in soil, and in oceanic and terrestrial subsurfaces, where the numbers of cells are 1.2 x 10(29), 2.6 x 10(29), 3.5 x 10(30), and 0. 25-2.5 x 10(30), respectively. The numbers of heterotrophic prokaryotes in the upper 200 m of the open ocean, the ocean below 200 m, and soil are consistent with average turnover times of 6-25 days, 0.8 yr, and 2.5 yr, respectively. Although subject to a great deal of uncertainty, the estimate for the average turnover time of prokaryotes in the subsurface is on the order of 1-2 x 10(3) yr. The cellular production rate for all prokaryotes on earth is estimated at 1.7 x 10(30) cells/yr and is highest in the open ocean. The large population size and rapid growth of prokaryotes provides an enormous capacity for genetic diversity.
Bacterial diversity and community structure in polygonal tundra soils from Samoylov Island, Lena Delta, Siberia
[J].
Soil bacterial community structure in Chinese wetlands
[J].Soil microbial communities are crucial in maintaining the functions of wetland ecosystems. Understanding the microbial community structure and the key factors driving the assemblages of wetland soil microbiota are important to reveal the connections between microorganisms and functions of wetland ecosystems. In this study, soil bacterial community compositions and the factors shaping them were investigated in three groups of wetlands across China, including Tibet plateau wetlands (TW), inland wetlands (IW) and coastal wetlands (CW). Overall, Bacterial community structure and turnover showed distinct patterns in different groups. Bacterial phyla were mainly composed of Proteobacteria followed by Chloroflexi, Acidobacteria, Actinobacteria and Bacteroidetes in all groups of wetland samples. At genus level, random forest model showed that Coprothermobacter and Acetobacter were two most important genera explaining the differences among groups. The abundances of these genera were very low in IW relative to the other two groups. The alpha diversity of IW was significantly higher than those of TW and CW. The relative contribution of environmental factors was larger in the assemblages of bacterial communities in TW and CW than that in IW. The pH and conductivity were recognized as the most important measured environmental factors influencing bacterial community structure. Our results suggested that the bacterial communities of wetlands in different regions were shaped with different mechanisms. The communities in CW and TW regions owned lower alpha diversity and were more influenced by deterministic processes than those in IW. In conclusion, the spatial pattern of soil bacterial community assembly in Chinese wetland was scale-dependent.
Bacterial community structure in two permafrost wetlands on the Tibetan Plateau and Sanjiang Plain, China
[J].Permafrost wetlands are important methane emission sources and fragile ecosystems sensitive to climate change. Presently, there remains a lack of knowledge regarding bacterial communities, especially methanotrophs in vast areas of permafrost on the Tibetan Plateau in Northwest China and the Sanjiang Plain (SJ) in Northeast China. In this study, 16S rRNA-based quantitative PCR (qPCR) and 454 pyrosequencing were used to identify bacterial communities in soils sampled from a littoral wetland of Lake Namco on the Tibetan Plateau (NMC) and an alluvial wetland on the SJ. Additionally, methanotroph-specific primers targeting particulate methane monooxygenase subunit A gene (pmoA) were used for qPCR and pyrosequencing analysis of methanotrophic community structure in NMC soils. qPCR analysis revealed the presence of 10(10) 16S rRNA gene copies per gram of wet soil in both wetlands, with 10(8) pmoA copies per gram of wet soil in NMC. The two permafrost wetlands showed similar bacterial community compositions, which differed from those reported in other cold environments. Proteobacteria, Actinobacteria, and Chloroflexi were the most abundant phyla in both wetlands, whereas Acidobacteria was prevalent in the acidic wetland SJ only. These four phyla constituted more than 80 % of total bacterial community diversity in permafrost wetland soils, and Methylobacter of type I methanotrophs was overwhelmingly dominant in NMC soils. This study is the first major bacterial sequencing effort of permafrost in the NMC and SJ wetlands, which provides fundamental data for further studies of microbial function in extreme ecosystems under climate change scenarios.
Soil pre-fumigation could effectively improve the disease suppressiveness of biofertilizer to banana Fusarium wilt disease by reshaping the soil microbiome
[J].
Sulfation of phenolic acids: chemoenzymatic vs. chemical synthesis
[J].
Soil acidification and the liming potential of biochar
[J].
Bacterial populations and environmental factors controlling cellulose degradation in an acidic Sphagnum peat
[J].Northern peatlands represent a major global carbon store harbouring approximately one-third of the global reserves of soil organic carbon. A large proportion of these peatlands consists of acidic Sphagnum-dominated ombrotrophic bogs, which are characterized by extremely low rates of plant debris decomposition. The degradation of cellulose, the major component of Sphagnum-derived litter, was monitored in long-term incubation experiments with acidic (pH 4.0) peat extracts. This process was almost undetectable at 10°C and occurred at low rates at 20°C, while it was significantly accelerated at both temperature regimes by the addition of available nitrogen. Cellulose breakdown was only partially inhibited in the presence of cycloheximide, suggesting that bacteria participated in this process. We aimed to identify these bacteria by a combination of molecular and cultivation approaches and to determine the factors that limit their activity in situ. The indigenous bacterial community in peat was dominated by Alphaproteobacteria and Acidobacteria. The addition of cellulose induced a clear shift in the community structure towards an increase in the relative abundance of the Bacteroidetes. Increasing temperature and nitrogen availability resulted in a selective development of bacteria phylogenetically related to Cytophaga hutchinsonii (94-95% 16S rRNA gene sequence similarity), which densely colonized microfibrils of cellulose. Among isolates obtained from this community only some subdivision 1 Acidobacteria were capable of degrading cellulose, albeit at a very slow rate. These Acidobacteria represent indigenous cellulolytic members of the microbial community in acidic peat and are easily out-competed by Cytophaga-like bacteria under conditions of increased nitrogen availability. Members of the phylum Firmicutes, known to be key players in cellulose degradation in neutral habitats, were not detected in the cellulolytic community enriched at low pH.© 2011 Society for Applied Microbiology and Blackwell Publishing Ltd.
Globally nitrogen addition alters soil microbial community structure, but has minor effects on soil microbial diversity and richness
[J].
Advances in actinomycete research: an ActinoBase review of 2019
[J].
Interactions of Bacillus spp. and plants—with special reference to induced systemic resistance (ISR)
[J].Biological control of soil-borne pathogens comprises the decrease of inoculum or of the disease producing activity of a pathogen through one or more mechanisms. Interest in biological control of soil-borne plant pathogens has increased considerably in the last few decades, because it may provide control of diseases that cannot or only partly be managed by other control strategies. Recent advances in microbial and molecular techniques have significantly contributed to new insights in underlying mechanisms by which introduced bacteria function. Colonization of plant roots is an essential step for both soil-borne pathogenic and beneficial rhizobacteria. Colonization patterns showed that rhizobacteria act as biocontrol agents or as growth-promoting bacteria form microcolonies or biofilms at preferred sites of root exudation. Such microcolonies are sites for bacteria to communicate with each other (quorum sensing) and to act in a coordinated manner. Elicitation of induced systemic resistance (ISR) by plant-associated bacteria was initially demonstrated using Pseudomonas spp. and other Gram-negative bacteria. Several strains of the species Bacillus amyloliquefaciens, B. subtilis, B. pasteurii, B. cereus, B. pumilus, B. mycoides, and B. sphaericus elicit significant reductions in the incidence or severity of various diseases on a diversity of hosts. Elicitation of ISR by these strains has been demonstrated in greenhouse or field trials on tomato, bell pepper, muskmelon, watermelon, sugar beet, tobacco, Arabidopsis sp., cucumber, loblolly pine, and two tropical crops (long cayenne pepper and green kuang futsoi). Protection resulting from ISR elicited by Bacillus spp. has been reported against leaf-spotting fungal and bacterial pathogens, systemic viruses, a crown-rotting fungal pathogen, root-knot nematodes, and a stem-blight fungal pathogen as well as damping-off, blue mold, and late blight diseases. This progress will lead to a more efficient use of these strains which is worthwhile approach to explore in context of biocontrol strategies.
Sphingomonas relies on chemotaxis to degrade polycyclic aromatic hydrocarbons and maintain dominance in coking sites
[J].
不动杆菌属分类的研究进展
[J].不动杆菌属是目前导致医院感染的常见细菌且耐药率很高,引起广泛关注。自1911年不动杆菌被分离鉴定以来,不动杆菌种属的分类和命名一直处于不断更新的状态。早期依据不动杆菌形态学和生化表型特征进行种属鉴定,但识别精度不高,经常出现菌种被错误鉴定的情况。随着分子生物学技术的发展,不动杆菌属的菌种可以通过全基因组测序技术被精确地鉴定和区分,这为深入研究不动杆菌的毒力和致病性差异提供了有力的支撑。本文对不动杆菌的分类历史和研究现状进行综述。
Progress of study on Acinetobacter classification
[J].Acinetobacter is a common bacterium that causes nosocomial infection at present, and the drug resistance rate is very high. Since the isolation and identification of Acinetobacter in 1911, the classification and naming of Acinetobacter species have been constantly updated.Early species identification based on morphological and biochemical phenotypic characteristics of acinetobacter was carried out, but the identification accuracy was not high, and the species were often wrongly identified. With the development of molecular biology technology, Acinetobacter species can be accurately identified and distinguished by whole genome sequencing technology, which provides a strong support for the in-depth study of the virulence and pathogenicity differences of Acinetobacter. This paper reviews the classification history and research status of Acinetobacter.
不同种植年限薰衣草根际土壤真菌群落结构的演变
[J].
Evolution of rhizosphere soil fungal community structure in different planting years of lavender
[J].
Soil potentials to resist continuous cropping obstacle: three field cases
[J].
Optimizing fertilizer management mitigated net greenhouse gas emissions in a paddy rice-upland wheat rotation system: a ten-year in situ observation of the Yangtze River Delta, China
[J].
Breeding beyond monoculture: putting the “intercrop” into crops
[J].
Can cereal-legume intercrop systems contribute to household nutrition in semi-arid environments: a systematic review and meta-analysis
[J].
Studies on the effect of different biofertilizers and organic manures on yeild and quality of strawberry (Fragaria ananasa duch.) cv. chandler under Allahabad agro climatic condition
[J].
Integrated instillation technology for the synthesis of a pH-responsive sodium alginate/biomass charcoal soil conditioner for controlled release of humic acid and soil remediation
[J].In this work, pH-responsive gel spheres for controlled release of humic acid (CSGCHs) were prepared by an integrated instillation technology using a composite material of sodium alginate (SA) and charcoal activated carbon (CAC) as a carrier, and their slow-release performance, pH-responsive performance, and soil amendment performance were investigated. The results showed that the prepared CSGCH was uniform in size with obvious base responsiveness. Soil remediation experiments revealed that CSGCH could play a good role in the remediation of different types of soils. After 50 days of remediation, the content of nutrients and organic matter in the soil increased significantly and the pH and salt content of saline soils decreased by 15.2 and 29.8%, respectively. The plant experiment showed that CSGCH could effectively promote the growth of crops. Therefore, the prepared soil conditioner has a great potential value for improving soil conditions and promoting crop growth in agricultural applications.
Microbiota composition and correlations with environmental factors in grass carp (Ctenopharyngodon idella) culture ponds in South China
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}